I - INTRODUCTION
Les spina bifida sont l’expression d’une dysraphie spinale. Au sens strict, ce terme désigne le défaut de fusion des lames vertébrales sur la ligne médiane ce qui peut entrainer une absence d’épineuse ou bien une épineuse bifide. Dans la pratique, ce terme a une signification beaucoup plus large puisqu’il inclue les myéloméningocèles et les méningocèles qui sont plus graves que le simple défaut de fusion des lames.
II - EMBRYOLOGIE
Les états dysraphiques sont la conséquence d’un trouble du développement de la gouttière neurale ou du tube neural. Les malformations qui en résultent peuvent donc affecter un ou plusieurs des tissus dérivant du neurectoderme : système nerveux, méninges, rachis, muscles paravertébraux, revêtement cutané. L’atteinte du système nerveux ou, au contraire, son intégrité conditionnent la gravité de ces états dysraphiques.
Pour la plupart des auteurs, les dysraphies spinales résultent d’un trouble de développement du neuropore postérieur, qu’il s’agisse soit d’un défaut de fermeture de la gouttière neurale vers la 3e, 4e semaine de la vie foetale, soit pour GARDNER de la réouverture d’un tube neural déjà formé sous l’effet d’une hydrocéphalie évolutive, et ceci vers le 2e, 3e mois du développement foetal.
III - CLASSIFICATION
Ces dysraphies regroupent de très nombreuses malformations habituellement classées en deux groupes : Les Spina Bifida Aperta (SBA) et Spina Bifida Occulta (SBO).
Il faut y adjoindre les fistules, les kystes dermoïdes, les kystes entériques et bronchogéniques.
Classiquement, les Spina Bifida Occulta (SBO) désignent la déhiscence d’une lame (C1, L5, S1 pour les plus fréquentes) découverte de façon fortuite sur une radiographie du rachis. Ces SBO sont asymptomatiques.
Par opposition, les Spina Bifida Aperta désignent les méningocèles et les myéloméningocèles.
Cette classification est abusive car stricto sensu le SBO devrait signifier que la malformation osseuse est insoupçonnable cliniquement. En fait il existe certains "SBO" qui s’accompagnent de stigmates cutanés très évocateurs du diagnostic.
IV - LES SPINA BIFIDA APERTA
Ce sont des malformations toujours diagnostiquées à la naissance et posant d’emblée un problème thérapeutique.
A. IncidenceEn France, l’incidence des SBA est de 0,5 o/oo environ, supérieure dans le Nord - Pas De Calais et la Bretagne (plus précisémment le nord finistère où elle atteint 2 o/oo).
L’incidence des SBA est plus élevée dans les pays anglo-saxons et notamment l’Irlande, l’Ecosse, ou le Pays de Galles (4/oo).
B. Epidémiologie Les enquêtes épidémiologiques montrent que la fréquence des SBA dépend au moins de trois facteurs :
1. Ethniques : Ces malformations sont moins fréquentes chez les Noirs et les Jaunes que chez les Blancs où elles affectent plus particulièrement les Anglo-Saxons
2. Génétiques : Elles sont plus fréquentes chez les filles que chez les garçons, et dans la famille d’un proposant, le risque chez un germain est grossièrement multiplié par 10
3. Exogènes : Variations séculaires de la fréquence des dysraphies ( 3 fois plus vers 1935 que vers 1965 à Boston ), et augmentation de leur incidence durant les mois d’hiver et dans les catégories socio-professionnelles défavorisées en Grande Bretagne. Ces facteurs exogènes consistent probablement en une carence vitaminique, notamment en acide folique
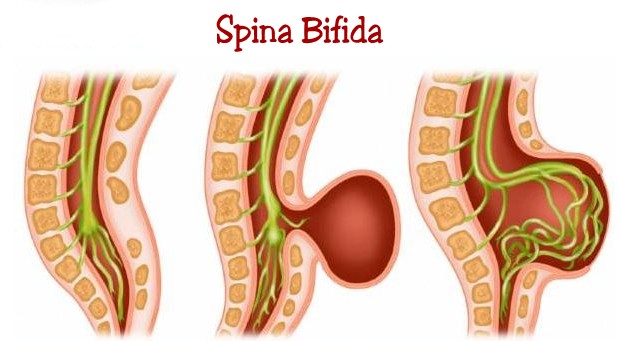
C. Formes anatomiquesDeux formes anatomiques sont à distinguer : la méningocèle et la myéloméningocèle car leur gravité est fondamentalement différente
1. La méningocèle (schéma 1A) : Il s’agit d’une hernie de la méninge seule , uniquement remplie de L.C.S.. Les éléments nerveux sont à leur place au fond du canal rachidien et en principe ne sont pas malformés. La hernie méningée fait saillie au travers de la brèche musculo-aponévrotique et osseuse du SB. Elle est responsable d’une tuméfaction médiane postérieure. Elle est habituellement recouverte de peau saine. Il s’agit ici de la forme bénigne des SBA ; mais celle-ci est malheureusement rare, ne représentant que 10 à 15 % des SBA .
2. La myélo-méningocèle (schéma 2) : Celle ci est beaucoup plus grave et malheureusement beaucoup plus fréquente.
Il y a hernie de tissu nerveux malformé dans la poche méningée. Le plus souvent, la moelle se termine à la face profonde du sac méningé en s’y étalant, prenant alors le nom de plaque médullaire. Les racines naissant de cette plaque sont malformées ; la moëlle en amont peut aussi être malformée ( syringomyélie, diastématomyélie). Habituellement, le recouvrement cutané de ces malformations n’est pas complet et est fréquemment remplacé par la dure-mère voire la seule arachnoïde. De la sorte, la moëlle est visible au dôme de la malformation. Celle-ci est fragile et laisse fréquemment suinter du L.C.S..
3. Le rachischisis : Le rachischisis est la forme extrème de la MM, et la plus grave. Ici, plus de sac ; la malformation n’est plus recouverte de méninges ; sur la ligne des épineuses, existe un large défect cutané et méningé permettant de voir la moelle anormale. Les corps vertébraux en regard sont également le siège de malformations majeures.
D. Anomalies associéesElles sont l’apanage presqu’exclusif des MM, et d’autant plus fréquentes que la MM est plus sévère et plus bas située.
1. L’ hydrocéphalie : Elle est présente dans 80 % des MM, et se développe dès l’age foetal comme le montrent la plupart des échographies anténatales de ces enfants. La macrocrânie est inconstante ; ce fait s’expliquerait par une fuite de L.C.S., soit intra amniotique chez le foetus, soit à l’air libre chez le nouveau-né. L’évolutivité de cette hydrocéphalie semble dépendre du siège de la malformation et surtout de la gravité du tableau neurologique. Ainsi, elle est plus fréquemment évolutive dans les formes lombaires (80 %) que dans les formes sacrées (50 %) ou thoraciques (43 %) ; elle est aussi d’autant plus fréquente que les déficits neurologiques sont plus graves : ainsi, l’hydrocéphalie est rare dans les meningocèles mais pratiquement constante dans les myéloméningocèles lombo-sacrées avec paralysies importantes. La cause habituelle de cette hydrocéphalie est la malformation d’ARNOLD CHIARI. Plus rarement, la cause en est une autre malformation : sténose de l’ Aqueduc de SYLVIUS, kyste de DANDY WALKER, kystes intra-cérébraux. Elle peut être enfin la conséquence d’une méningite consécutive à l’infection du MM. Il n’est pas rare que cette hydrocéphalie puisse se stabiliser spontanément. Inversement, une hydrocéphalie apparemment stabilisée peut se décompenser dans les suites de la fermeture de la myéloméningocèle. Une des raisons invoquées est la suppression de la fuite de L.C.S. à ce niveau.
2.. La malformation d’ARNOLD CHIARI : Elle se définit par la présence anormale dans le rachis cervical des amygdales cérebelleuses, parfois d’une partie des hémisphères cérébelleux et du tronc cérébral.
Par ordre de gravité on a décrit les formes 1 ( hernie exclusive des amygdales), 2 (hernie des amygdales et du tronc +/- du cervelet), 3 ( encéphalocèle occipitale associée à une forme 2).
Dans la MM, la forme habituelle est le CHIARI 2. Il est à noter que dans les cas de SBA, la malformation d’ARNOLD CHIARI ne s’associe pas à des malformations de la charnière cervico-occipitale.
3. Ses conséquences : Ses conséquences possibles sont au nombre de 4. Elles peuvent éventuellement s’ associer entre elles . Par ordre décroissant de fréquence, elles sont :
l’ hydrocéphalie, pratiquement constante, par blocage du L.C.S. au niveau des orifices de sortie du IVe ventricule et/ou des espaces sous-arachnoïdiens du rachis cervical haut et de la base du crâne.
la paralysie plus ou moins importante des paires crâniennes basses ( IX, X, XI ; parfois XII), responsable de stridor par paralysie des dilatateurs de la glotte, de troubles de la déglutition, de troubles de la phonation. Ces paralysies peuvent être déclenchées, aggravées ou entretenues par la survenue de l’hydrocéphalie.
Des signes de compression de la moelle cervicale haute se traduisant par un syndrome pyramidal des membres supérieurs et parfois des déficits moteurs au même niveau.
Eventuellement une syringomyélie et/ou une syringobulbie.
Autres malformations : il faut rechercher l’existence d’une agénésie du corps calleux et celle d’une petite fosse postérieure avec implantation très basse des sinus latéraux, conséquence de la malformation d’ARNOLD CHIARI.
E. Examen clinique d’un nouveau né porteur d’un SBAL’examen doit se fixer 4 objectifs :
- reconnaître la malformation
- en apprécier les conséquences au niveau des membres inférieurs, des sphincters, et de l’encéphale
- rechercher des malformations associées
- dresser un pronostic fonctionnel.
1. Reconnaître la malformation : Le plus souvent, le diagnostic est évident : tuméfaction de volume variable, implantée plus ou moins largement sur la ligne médiane, habituellement dans la région lombo-sacrée, plus rarement dans la région cervicale, exceptionnellement au niveau dorsal. Selon le type de la malformation, le revêtement cutané est soit complet (M) soit incomplet (MM). Dans ce dernier cas, le dôme de la malformation est occupé par un tissu charnu rosé et suintant ( la plaque médullaire) ; de part et d’autre de la plaque, le revêtement de la poche est translucide ( arachnoïde, dure mère) jusqu’à parvenir en périphérie à l’épithélium normalement constitué.
Devant un SBA, il faut :
- apprécier la taille de la malformation
- apprécier la qualité du revêtement cutané
- rechercher une fuite de L.C.S. (suintement au niveau de l’aire médullaire ou écoulement massif de L.C.S. parfois responsable de l’affaissement du sac méningé)
Le diagnostic clinique entre méningocèle et myéloméningocèle est en règle simple, la première, au contraire de la deuxième, se caractérisant par un examen neurologique normal et une malformation épithélialisée.
Dans de rares cas cependant, la MM peut être recouverte de peau, soit d’emblée, soit du fait d’une épithélialisation secondaire.
La M peut aussi s’accompagner de déficits. C’est pourquoi des examens neuro-radiologiques s’imposent pour en déterminer le type.
2. Apprécier les conséquences au niveau des membres inférieurs, des sphincters, et de l’encéphaleMembres inférieurs. On appréciera la qualité de la gesticulation spontanée de l’enfant, sa réactivité à la douleur, et la présence de signes indirects de paralysie : déformation du squelette jambier, pied-bot, amyotrophie des membres et des fesses, et flessum de hanche.
Sphincters : On recherchera :
soit des signes évidents de paralysie sphinctérienne : fuite urinaire permanente ou lors des cris et à la verticalisation, béance anale, voire prolapsus
soit des signes plus discrets ; leur détection est alors difficile et ne s’apprécie vraiment que par l’examen de l’anus : disparition des plis radiaires de la marge anale, absence de réflexe anal, hypotonie du sphincter anal et du périnée.
Crâne : L’objectif est de rechercher une hydrocéphalie et les conséquences possibles d’une éventuelle malformation d’ARNOLD CHIARI. Il faut donc apprécier la taille et la tension des fontanelles, mesurer le périmètre crânien, rechercher un stridor, des troubles de la déglutition et des réflexes pyramidaux aux membres supérieurs en se rappelant qu’un périmètre cranien normal ou bas n’élimine pas l’existence d’une dilatation ventriculaire.
Articulation de la hancheà la recherche d’une luxation
3. Rechercher des malformations associées. Celles-ci sont présentes chez environ 10% des enfants atteints. L’étude de grandes séries d’enfants avec SBA a montré une moyenne de 2,2 malformations associées par patient. Une telle fréquence rend donc absolument nécessaire la pratique d’un examen somatique clinique et radiologique complet en vue de leur dépistage.
4. Dresser un pronostic fonctionnel. Hormis les cas extrèmes (déficits massifs ou en apparence nuls), il est toujours très difficile d’estimer ce que sera le devenir fonctionnel de l’enfant. Schématiquement, il faut opposer les malformations sacrées, responsables de troubles sphinctériens exclusifs, à celles plus haut situées qui, en outre, entrainent des paralysies de gravité variable au niveau des membres inférieurs. Parmi ces dernières, il faut distinguer celles où les paralysies incluent les fessiers et le quadriceps de celles où les paralysies sont essentiellement distales. Les premières vont entraver la station debout et nécessiter de grands appareillages pelvi-cruro-pédieux avec aide de marche, voire le fauteuil roulant. Les secondes nécessiteront des appareillages plus légers, ou de simples chaussures orthopédiques. Cependant, quant l’atteinte des fessiers est importante (S1), l’érection du tronc est compromise pouvant nécessiter l’utilisation de cannes ou d’appareillage lourd.
F. Examens complémentairesCertains sont à faire d’emblée :
1-radiographies du rachis et du thorax pour préciser le niveau et l’étendue du SB et dépister d’autres malformations
2-radiographies des hanches à la recherche d’une luxation
3-radiographies des pieds en charge4-radiographies du crâne 5-une échographie et/ou un scanner ou I.R.M. cérébralpour apprécier le degré de dilatation des ventricules et l’existence éventuelle d’une malformation cérébrale associée
6-Une I.R.M. médullaire quand la malformation est épithélialisée ou de siège dorsal ou cervical.
D’autres seront à faire secondairement, après l’intervention sur la malformation :
une échographie rénale et vésicale +/- une urographie intra veineuse avec cystographie rétrograde pour connaître l’état des voies excrétrices urinaires et rechercher résidu vésical et reflux vésico-urétéral
![]() enfin, en cas de stridor, de paralysies de la déglutition, ou de troubles neurologiques des membres supérieurs, il peut être utile de demander une laryngoscopie pour préciser l’état de motricité des cordes vocales, et une I.R.M. de la charnière cranio-cervicale pour apprécier la gravité de la malformation d’ARNOLD CHIARI.
enfin, en cas de stridor, de paralysies de la déglutition, ou de troubles neurologiques des membres supérieurs, il peut être utile de demander une laryngoscopie pour préciser l’état de motricité des cordes vocales, et une I.R.M. de la charnière cranio-cervicale pour apprécier la gravité de la malformation d’ARNOLD CHIARI.
G. Conduite à tenir 1. En période prénataleDans la maternité...Il faut préserver la lésion de toute souillure et éviter sa rupture. Pour ce faire, placer l’enfant en décubitus ventral, recouvrir le SB d’un pansement stérile, gras ou humide, de compresses puis d’un pansement occlusif et épais.
Dans le service spécialisé...L’enfant doit être pris en charge par une équipe pluridisciplinaire, dont le rôle est de faire le bilan des lésions et de porter l’indication opératoire. En théorie, il faut opérer d’urgence les malformations rompues, opérer rapidement celles qui menacent de se rompre, opérer de façon différée celles qui sont épithélialisées ( à l’abri d’une rupture et donc de méningite).
En pratique, les indications sont parfois modulées en fonction de la gravité des déficits et de la volonté des parents ; de ce fait, dans certains centres, l’abstention chirurgicale en période néo-natale est devenue plus fréquente, le traitement de la malformation étant alors retardé de quelques semaines pour ne s’appliquer qu’aux survivants. La cure chirurgicale de la malformation ne peut en principe espérer améliorer les paralysies existantes ; elle vise à réintégrer dans le canal rachidien les éléments nerveux éventuellement contenus dans la poche puis à fermer de façon étanche les méninges et la peau (figure 1B).
Le traitement chirurgical de la myélo-méningocèle ne résume pas les soins à apporter à l’enfant. Il faut en outre :
![]() traiter l’hydrocéphalie quand elle est évolutive, par la mise en place d’une valve.
traiter l’hydrocéphalie quand elle est évolutive, par la mise en place d’une valve.
![]() prévenir les déformations articulaires ou les corriger par une kinésithérapie et la confection d’attelles de posture et/ou coussins d’abduction
prévenir les déformations articulaires ou les corriger par une kinésithérapie et la confection d’attelles de posture et/ou coussins d’abduction
![]() prévenir ou déjà traiter les infections urinaires en instaurant, à condition qu’il n’y ait pas de reflux vésico-urétéral, des expressions vésicales pluriquotidiennes +/- antibiotiques. En cas de reflux, on pratiquera des sondages vésicaux intermittents.
prévenir ou déjà traiter les infections urinaires en instaurant, à condition qu’il n’y ait pas de reflux vésico-urétéral, des expressions vésicales pluriquotidiennes +/- antibiotiques. En cas de reflux, on pratiquera des sondages vésicaux intermittents.
2. Traitement à long terme...Il nécessite la collaboration d’une équipe multidisciplinaire. Il a pour objectif d’éviter, de dépister ou de traiter trois types de complications :
Orthopédiques...Avant tout les déformations articulaires, parfois les troubles de la statique rachidienne, les fractures pathologiques, les maux perforants
Urologiques...Infections urinaires, reflux urétéral, hydronéphrose, lithiases
Neurochirurgicales...Infection ou obstruction de valve, décompensation tardive de la malformation d’ARNOLD CHIARI, ou beaucoup plus rarement aggravation neurologique que pourraieny entraîner une syringomyélie, une diastématomyélie ou une moelle fixée.
La prise en charge de ces patients nécessite en outre la présence de rééducateurs, de kinésithérapeutes, d’éducateurs, et de psychologues.
H. RésultatsCette prise en charge pluridisciplinaire a permis une amélioration considérable du sort de ces enfants. Les handicaps majeurs restent les troubles moteurs et l’incontinence sphinctérienne.
Sur le plan moteur, la majorité des enfants auront des appareillages et subiront des interventions orthopédiques correctrices multiples. Certaines séries font état de 32% de sujets autonomes avec ou sans appareillage.
Sur le plan sphinctérien, il faut distinguer d’un point de vue social les enfants dont la paralysie vésicale est flasque de ceux dont la paralysie vésicale est spastique. Les premiers sont en effet incontinents , ce qui gène leur intégration sociale. Les seconds peuvent être propres à condition que la vessie soit vidée à intervalles fixes par sondages. Chez l’enfant de plus de 5-6 ans, il peut s’agir d’auto-sondages. La pratique de ces sondages et, chez les enfants incontinents les expressions vésicales pour vider la vessie le plus complètement possible ont permis de réduire considérablement les indications de dérivations urinaires à la peau ainsi que la fréquence des infections urinaires. L’insuffisance rénale est devenue l’exception.
Plus de 70% des malades ont des possibilités intellectuelles leur permettant une scolarité ou un apprentissage normaux.
Le traitement d’un SBA est lourd. Un enfant valvé nécessite une moyenne de 3 interventions chirurgicales ; 80% des enfants subissent entre quatre et six interventions orthopédiques et 41% au moins une intervention urologique.
I. Conseil génétique, diagnostic anténatal et prévention1. Le depistageLe véritable "traitement" d’un SBA est préventif soit par détection anténatale et interruption thérapeutique de grossesse, soit prophylactique.
Le diagnostic antenatal se fait par :
L’échographie foetale Elle montre typiquement le défect osseux, l’ectasie méningée, l’hydrocéphalie, la petite fosse postérieure et souvent des pieds bots et des membres inférieurs peu mobiles.
L’amniocentèse Elle permet le dosage à la 16e semaine de grossesse de l’alpha-foeto-protéine et de l’acétylcholinestérase dans le liquide amniotique . Leur augmentation n’existe cependant que dans les malformations ouvertes.
L’I.R.M. Si l’échographie fait partie du bilan systématique de surveillance d’une grossesse, l’amniocentèse est réservée aux grossesses à risque ( premier enfant atteint de SBA) et aux foetus avec anomalies échographiques, et l’I.R.M. est indiquée en cas d’échographie douteuse. Il faut savoir que l’échographie peut être apparemment normale dans les formes mineures de SB.
2. La préventionDes études récentes ont démontré qu’un régime riche en acide folique administré à titre prophylactique pendant la période péri-conceptionnelle aux femmes à risque ou originaires des régions ou les SBA sont particulièrement fréquents aboutissait à une diminution significative de l’incidence de ces malformations.
V - LES SPINA BIFIDA OCCULTA ( figure 3)
Ce terme est surtout radiologique et concerne le manque d’épineuse ou de lame habituellement en C1, L5, ou S1 sur un ou deux niveaux. Ces "SBO" sont fréquents (10 à 20 % dans la population normale). En fait, on englobe aussi dans les SBO des malformations plus complexes avec SB plus étendu, malformation du système nerveux et hamartome.
On doit donc distinguer :
les spina bifida occulta simples, isolés,
les spina bifida occulta complexes
Parmi ces derniers, les plus fréquents sont les lipomes et les fistules.
A. Les spina lipomes 1. Description anatomique Il s’agit de lipomes intra-rachidiens avec anomalies rachidiennes et nerveuses, et parfois d’autres malformations associées.
Le lipomeIl s’agit typiquement d’une masse graisseuse à développement typiquement extra- et intra-rachidien. La masse extra- rachidienne est présente trois fois sur quatre et se continue sans limite avec le pannicule adipeux normal ; elle déforme donc le revêtement cutané sus jacent. En revanche, la masse intra-rachidienne est constante et pénètre la dure-mère et l’arachnoïde du cul-de-sac lombaire en se fixant sur la moëlle avec une interface fibreuse entre les tissus nerveux et graisseux. Trois fois sur quatre, le lipome s’insère sur la face postérieure de la moëlle ou en termino-terminal sur le cône ; les racines naissent alors en avant du lipome et sont donc extra-lipomateuses. Une fois sur quatre, le lipome s’insère sur les faces postérieure et latérale de la moëlle et les racines ont un trajet intra-lipomateux. Cette distinction est importante à faire d’un point de vue chirurgical : si les interventions sont faciles dans le premier cas, elles sont difficiles dans le second.
Les anomalies rachidiennesLe spina bifida est constant, prédominant nettement au niveau lombaire et sacré, et étendu au moins à deux niveaux. Il peut s’y associer d’autres anomalies vertébrales.
Les anomalies nerveusesIl existe de façon quasi-constante une moëlle basse, se terminant le plus souvent en L4 L5 ou au dessous, avec par voie de conséquence une perte de la disposition en queue de cheval des racines et d’une direction non plus descendante, mais horizontale ou ascendante de celles-ci. Il peut exister d’autres anomalies comme l’hydromyélie (25%), ou de malformations plus ou moins complexes des racines.
Autres malformationsLes plus fréquentes (95%) sont cutanées en regard du lipome, avec la présence d’une masse sous cutanée, d’une ombilication, ou d’un angiome qui sont les plus fréquentes. Ces malformations ont une importance diagnostique capitale.
2. Conséquences physiopathologiquesLa moëlle fixéeLa moëlle est amarée à la peau, au tissu sous-cutané et aux méninges par l’intermédiaire du lipome
La moëlle tendueConséquence directe de sa fixation, cette tension de la moëlle peut être la cause de troubles neurologiques apparaissant progressivement.
La moëlle compriméeLe volume du lipome peut être tel qu’il peut agir sur la moëlle à la manière d’une tumeur extra-médullaire. Quelques cas de compression médullaire ont été décrits à l’occasion du développement d’une obésité.
3. Circonstances de découverteIl existe grossièrement deux types de circonstances de découverte : chez le nouveau-né ou nourrisson avec des anomalies cutanées, ou bien chez l’enfant, adolescent ou adulte chez qui se développent progressivement des troubles neurologiques ou orthopédiques.
Les anomalies cutanéesIl s’agit la plupart du temps (75%) d’une voussure des parties molles médiane ou paramédiane pouvant donc entrainer une déviation du pli fessier. Les autres anomalies sont un angiome, une hypertrichose, une zone d’aplasie cutanée, une ombilication cutanée. Dans 5% des cas seulement il n’existe pas d’anomalies cutanées.
Les troubles neurologiquesIls consistent essentiellement en des troubles sphinctériens ( pollakiurie, dysurie, mictions impérieuses avec fuites) , constipation, et aux membres inférieurs des déficits mixtes et distaux ( pieds creux, amyotrophie d’un mollet ...).
Ces troubles sont souvent congénitaux ; parfois ils sont acquis et évolutifs. Ils sont d’autant plus souvent observés que le sujet est plus agé.
4. Examens complémentairesRadiographies standard du rachisLes anomalies sont constantes mais peuvent être difficiles à détecter chez le nouveau né et le nourrisson. Elles consistent en un spina osseux étendu à au moins 2 niveaux, parfois en une agénésie ou hémi-agénésie sacrée voire à des malformations vertébrales plus complexes.
I.R.M.C’est l’examen nécessaire et suffisant à affirmer le diagnostic en montrant un hypersignal sur les séquences pondérées en T1 (signal de la graisse) Il permet en outre d’apprécier la configuration du lipome et en particulier sa topographie par rapport à la moëlle.
Bilan urodynamique et electromyogrammeCes examens ont un intérêt pronostique et de référence pour la surveillance ultérieure.
5. TraitementPrincipesIl s’agit de libérer la moëlle et si besoin de la décomprimer. Les interventions ne peuvent se faire que sous microscope. Pour certains, elles nécessitent l’utilisation de stimulations électriques per opératoires pour repérer les racines
RésultatsAu plan anatomique, et dans 1/4 des cas, la libération médullaire est incomplète du fait de la complexité de la malformation.
Au plan fonctionnel, le risque d’aggravation neurologique post opératoire est de 4%.
A long terme, 9% des opérés continuent à s’aggraver ou développent des troubles en dépit de l’intervention. Ce chiffre est à mettre en balance avec 50- 60% d’aggravation neurologique spontanée. 67% des opérés s’améliorent. La guérison neurologique est exceptionnelle.
IndicationsIl existe deux attitudes : la première est de surveiller les patients asymptomatiques et de ne leur proposer une intervention que lorsque les troubles neurologiques apparaissent ; la seconde est d’intervenir préventivement et précocément. C’est l’attitude actuellement pronée.
B. Les fistules Elles sont le plus souvent lombaires et sont révélées par l’examen clinique, par la survenue de méningites à répétition, ou par l’apparition d’une paraplégie d’évolution rapide ( ce dernier cas devant avant tout faire évoquer la possibilité d’un abcès ).
Anatomiquement, elles sont soit borgnes, et alors bénignes, soit ouvertes dans la dure mère, et éventuellement prolongées par un kyste dermoïde qui peut être intra- ou extra- médullaire. Devant la découverte d’une fistule, il faut donc faire en urgence une I.R.M.. De même, les méningites à répétition doivent faire rechercher une fistule.
Le traitement ne doit jamais se cantonner à l’exclusion seule de la fistule, mais poursuivre celle-ci jusqu’à son point de pénétration durale, en poursuivant par l’ouverture systématique de la dure mère et l’ablation éventuelle d’un kyste dermoïde.
C. Les fistules sacro-coccygiennesCes fistules sont à distinguer des fistules sacro-coccygiennes qu’il conviendrait mieux d’appeler "fossettes coccygiennes" .
Ici, la fistule cutanée se prolonge par un tractus fibreux qui s’insère à la pointe du coccyx.
Ces fistules sont fréquentes et d’une grande bénignité ; (il n’y a pas de dure mère dans le coccyx ,ni de spina bifida ). Le seul risque est la surinfection locale et la formation d’un kyste pilonidal.
Dans quelques cas cependant, elles peuvent s’associer à des anomalies de la moëlle terminale.
Le traitement peut être soit l’abstention avec une hygiène cutanée, soit la chirurgie par excision de la fistule à titre prophylactique.
LEXIQUE DES ABREVIATIONS UTILISEES :
SB : Spina bifida
SBA : Spina bifida aperta
SBO : Spina bifida occulta
M : Méningocèle
MM : Myéloméningocèle
BIBLIOGRAPHIE
1 - Briard ML, Le Merrer M, Frezal J : Epidémiologie des malformations du tube neural. Gaz. Med. de France 89 : 3075-3079, 1982
2 - Carmel PW : The Arnold Chiari malformation. Pediatric Neurosurgery. Surgery of the developping nervous system. Grune and Stratton, 1982, pp 9-77
3 - Epstein F et al : Delayed cauda equina reconstruction in myelomeningocele
Neurosurgery 6 : 540-541, 1980
4 - Genevet L : Devenir à long terme des myeloméningocèles. La Rev Ped 17 : 523-528, 1981
5 - Hirsch JF et al : Lumbosacral lipomas with spina bifida. Child’s Nerv Syst 4 : 354-360, 1988
6 - Mac Lone DG, Knepper PA : The cause of Chiari 2 malformation : a unified theory. Pediatr Neurosci 15 : 1-12, 1989
7 - Pierre-Kahn A et al : Intraspinal lipomas with spina bifida. J Neurosurg 65 : 756-761, 1986
8 - Pierre-Kahn : Les spina lipomes. Arch Fr Pediatr 48 : 45-51, 1991









 Français
Français
 English
English
 Español
Español
 Italiano
Italiano
 Deutsch
Deutsch
 Nederlands
Nederlands
 Portuguesa
Portuguesa





 ème visiteur
ème visiteur